 Associate Professor Matthew Simunovic is a Sydney-based ophthalmologist who specialises in medical retina and vitreoretinal surgery and is also involved in research.
Associate Professor Matthew Simunovic is a Sydney-based ophthalmologist who specialises in medical retina and vitreoretinal surgery and is also involved in research.
He graduated from medical school at the University of Cambridge, where he also obtained his PhD and aspired to become an ophthalmologist. Prior to this, he studied optometry at UNSW with his love for eyes stemming from a young age by his uncle who was an optometrist in Brisbane. He completed his fellowship training at The University of British Columbia in Vancouver, Moorfield’s Eye Hospital in London and at the John Radcliffe Hospital/Nuffield Laboratory of Ophthalmology at the University of Oxford.
A/Prof. Simunovic is currently working in the retinal unit at Sydney Eye Hospital and the Sydney Children’s Hospital network. He works in private practice at Retina Associates Bondi Junction and Chatswood as well as Sutherland Eye Surgeons and operates privately at Bondi Junction Private Hospital. He is an Associate Professor at the Save Sight Institute at the University of Sydney and his research group conducts clinical research and laboratory research into developing treatments for retinal disease, improving visual function as well as experimental vitreoretinal surgery. His research is currently being supported by the Macular Diseases Foundation Australia and the NHMRC.
To see a list of the research projects and publications A/Prof. Simunovic is involved in, click here.
If any YO members are interested in being involved with research with A/Prof. Simunovic, send us an email to get in contact!
Read the interview with A/Prof. Simunovic below for a detailed description on his recent clinical research and the life-changing impact that Luxturna is having on patients with Leber’s Congenital Amaurosis 2.
LUXTURNA
What is Luxturna?
Luxturna (voretigene neparvovec) is the first ever ocular gene therapy recently approved here in Australia by the Therapeutics Goods Administration (TGA) for use in patients with Leber’s Congenital Amaurosis 2 (LCA2). LCA2 is a retinal dystrophy caused by biallelic mutations to the RPE65 gene, which is essential in recycling the chromophore (the modified version of Vitamin A bound to rhodopsin and the cone opsins). In patients with LCA2 where there is no functional RPE65 in the eye but the RPE and photoreceptors are still intact, the introduction of the RPE65 gene results in the production of the RPE65 protein, establishing the visual cycle and improving vision.
Who and what did the research involve?
The research which led to the development of Luxturna was mainly conducted in the USA, initially developed in the laboratory of Prof. Jean Bennett at the University of Pennsylvania.
The research team involved in delivering the treatment here in Australia commenced with identification and genotyping patients, which was performed by my colleagues Prof. Robyn Jamieson and Prof. John Grigg. Preparation of the medication according to the regulations surrounding genetically modified organisms (GMOs) was led by Peter Barclay in the pharmacy at Children’s Hospital Westmead. A theatre nursing team, anaesthetists and recovery teams were involved, as well as the surgical team which included myself and Dr Gaurav Bhardwaj.
Novartis supply and distribute Luxturna and were primarily responsible for regulatory approval in Australia.
What did the surgery involve?
The surgery initially involves a small gauge pars plana vitrectomy, where three small, valved ports are inserted through the sclera anteriorly to the retina via the pars plana. Once inserted, an infusion line is run into one of the ports. This provides a steady flow of balanced salt solution (designed to mimic aqueous humour) to prevent the eye from collapsing when the vitreous is cut and aspirated. The vitreous is removed using a vitrector through another port (a miniscule instrument with 0.5mm diameter that has a guillotine cutter and aspiration line). The final port allows for a light pipe into the eye so that the fundus is viewed via an indirection viewing system at a surgical microscope. In many patients, a posterior vitreous detachment needs to be induced surgically, which can be quite tricky in younger patients and children with firmer vitreoretinal adhesions.
Once the bulk of the core and peripheral vitreous has been removed, the sub-retinal injection is performed using a 38-guage Teflon tipped cannula (0.125mm diameter) placed in the subretinal space. Once in the correct surgical plane, the injection commences, however most surgeons prefer to create a bleb first using a balanced salt solution before the injection so that they can use the on-table OCT to confirm they are in the correct plane. Inadvertently injecting Luxturna into the choroid or the vitreous can potentially lead to lack of efficacy and/or inflammation. Once in the correct surgical plane, a total of 0.3mL Luxturna is injected via the same puncture site into the bleb and the surgeon will then check for peripheral retinal holes or tears using 360-degree scleral indentation (treated with cryopexy or laser if they do arise). The fluid is then exchanged in the vitreous cavity for filtered air to encourage migration of subretinal fluid to the desired treatment area during post-operative positioning. It also assists in the closure of the scleral incision sites/assessing the patency of closure as well as helps prevent retinal detachment if any retinal holes or tears are found.
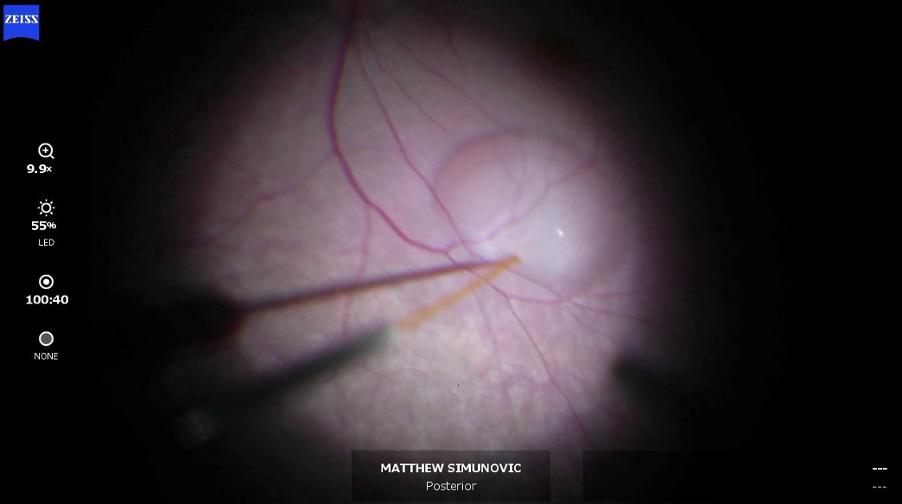
Image: The Luxturna sub-retinal injection in action.

Image: A/Prof. Simunovic operating on the first patient to be treated with Luxturna
Once the surgery is complete, the patient is positioned in a way that allows for gravity to move the Luxturna to the position of the residual island of viable retina. This area is usually the macula. For example, if the injection is performed superior to the macula, the patient may be positioned upright or face up.

Image: The Research Team (from left to right): Prof. John Grigg (paediatric ophthalmologist), Prof. Robyn Jamieson (clinical geneticist), Dr Gaurav Bhardwaj (vitreoretinal surgeon), A/Prof. Matthew Simunovic, Nicole Johnston (Novartis), Ajit Viswalingam (Novartis), Sonja Nikolic (Novartis)
Since a pars plana vitrectomy is involved, what is the risk of cataract formation in these patients?
While it is well-known that pars plana vitrectomy accelerates nuclear sclerosis and cataract formation, there are some patients that are resilient to its effects. This includes younger patients and diabetics. To date, some of the older patients have had pre-existing cataract, which is anticipated to progress more rapidly following surgery. However, cataract extraction is not performed concurrently for Luxturna patients.
Did the treatment in these cases work as was hypothesised?
Improvements in key functional tests have been seen in treated patients which mirror those of the Phase III/pivotal trials. The latter demonstrated a 2.1 log unit improvement (21dB) in retinal sensitivity, which has translated to significant real-world improvements. For example, the first patient treated saw me recently and mentioned that they’d seen stars for the first time. There have also been subjective improvements in colour vision, form discrimination and near vision, with one patient now about to crochet and thread larger needles. Treatment takes about 2 weeks to work, with one patient no longer using a mobility cane to navigate under street lighting about 2 weeks following their surgery.
What inspired you to get involved with research within genetic disorders?
I did my PhD at a time when there was a whirlwind of activity in the area of molecular biology for inherited retinal disease and the vague hope that gene therapy was on the horizon. Of course, even back then, there were important things that clinicians could do to help patients, but it was exciting to know that a common (1 in 2,000-3,000 and the commonest cause of blindness in children and those of working age) group of conditions might one day be “treatable”. Likewise, it has been exciting to be involved in gene therapy and other emerging treatments, not least because they offer hope of meaningful improvements in vision and quality of life for patients and their families.
What is next for you in terms of research/work?
We have more patients coming for Luxturna surgery in the New Year. On the clinical research front, my team and I are involved in clinical trials of therapeutics for retinal detachment, age-related macular degeneration and macular telangiectasia type II. We are also conducting an investigator-initiated trial looking at the fate of subretinally injected drugs using a fluorophotometry protocol we have developed. My group is also conducting clinical projects exploring visual function and structure in retinal disease (including inherited retinal disease and surgical retinal disease) which draws on machine-learning (in collaboration with colleagues in Computer Science at USyd). In addition, we are in the throes of completing an inherited retinal disease registry, which the Save Sight Registries host. This has been a significant undertaking with input from clinicians in Australia, New Zealand, the USA, Germany, Switzerland, Belgium, Portugal and the UK. Finally, in the lab, we are developing causative mechanism independent approaches to treat retinal degeneration using different optogenetic approaches – one approach is targeted at addressing vision loss from dry ARMD (funded by the Macular Disease Foundation Australia), and the other addresses inherited retinal degeneration of differing severity (supported by the NHMRC).
How does this research apply to optometrists?
Optometrists see patients with inherited retinal degeneration both at initial presentation and for ongoing care. Therefore, they are well-placed to keep patients informed about developments in the area and provide appropriate referrals. Many patients with these conditions have been told that “nothing can be done”. Even before the advent of gene therapy, this was not true. This group of patients benefit from refractive management and often from low vision aids and active management of associated problems like cataract, cystoid maculopathy, epiretinal membrane, etc. It’s crucial that patients are offered the chance to be assessed by clinicians with an interest in inherited retinal disease and by a genetics service, whether the patient wishes to undergo “genetic testing” or not.
